我院高威帷副教授、赵纪军教授以及德州大学奥斯汀分校James R. Chelikowsky教授,在Physical Review Letters上发表重要工作《Efficient Full-Frequency GW Calculations Using a Lanczos Method》(Lanczos加速的高效全频率GW计算),被选为编辑推荐工作。

电子结构是决定物质性质的根本因素之一。在计算物理学领域,准确理解和高效地计算物质的电子结构是一个基本问题和挑战。
密度泛函理论(Density Functional Theory, DFT)是目前物质科学中计算电子结构的主要方法。但是,DFT也存在明显的局限性,例如无法给出准确的半导体能隙等关键物理量。在半导体光伏材料能隙的模拟和设计中,即使计算结果与真实值相差0.5电子伏特,也会导致对材料性能描述的巨大偏差。
基于量子多体微扰理论的GW近似方法提供了一种相比密度泛函理论更精确可靠的电子结构计算方法,但是, GW近似对计算资源的需求也远高于DFT,计算复杂度一般大于O(N3)。因此,受限于计算资源,GW近似方法一般用于包含少于100原子的物质体系,很难应用于精确计算大尺寸复杂体系。
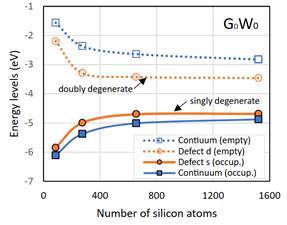

基于上述问题,高威帷副教授、赵纪军教授、德州大学奥斯汀分校唐昭博士、以及James R. Chelikowsky教授合作,开发了一类基于Lanczos算法的理论,极大提升了GW计算的效率(相比原方法提升5到6个数量级),使得研究者可以在较小计算资源消耗下,实现几百到数千原子的精确电子结构计算效率。该工作为人们精确计算大尺寸的复杂物质体系的电子结构和激发态性质提供了新途径。
该工作中,高威帷副教授为第一作者,赵纪军教授和James R. Chelikowsky教授为通讯作者,成果部分得到大连理工大学基本科研业务费的资助。
论文链接:
Phys. Rev. Lett. 132, 126402 (2024);
其他相关工作
Electronic Structure 4 (2), 023003 (2022); DOI:10.1088/2516-1075/ac709a
J. Chem. Theory Comput 16, 4, 2216 (2020);
